Phrynosoma blainvillii (Blainville’s Horned Lizard)
Visual overview of genome assembly metrics
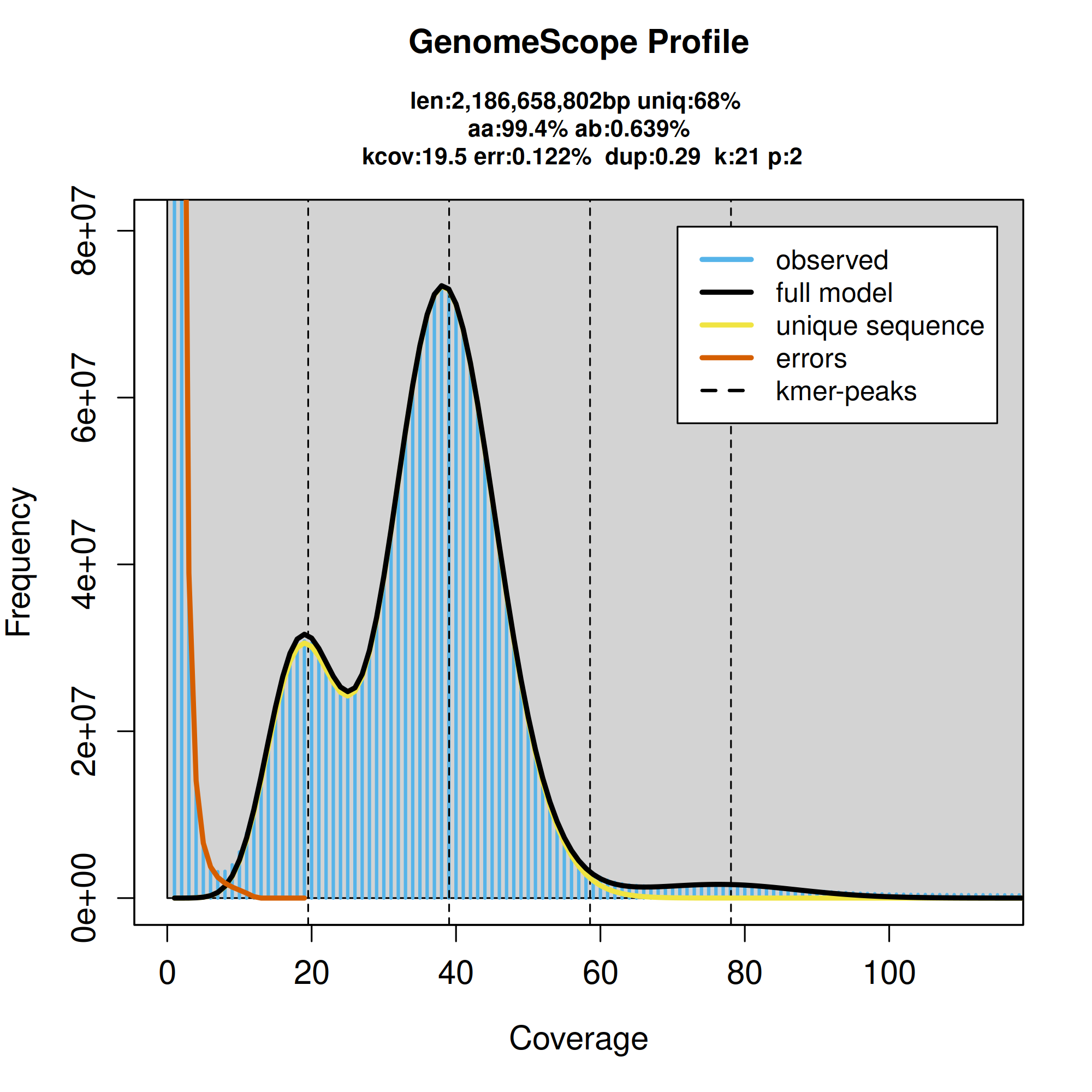
K-mer spectra output generated from PacBio HiFi data without adapters using GenomeScope2.0. The bimodal pattern observed corresponds to a diploid genome. K-mers covered at lower coverage and low frequency correspond to differences between haplotypes, whereas the higher coverage and high frequency k-mers correspond to the similarities between haplotypes.
BlobToolkit Snail plot displaying the quality metrics for the P. blainvillii primary assembly (haplotype 1; rPhrBla1). The plot circle represents the full size of the assembly. From the inside-out, the central plot covers length-related metrics. The red line represents the size of the longest scaffold; all other scaffolds are arranged in size-order moving clockwise around the plot and drawn in gray starting from the outside of the central plot. Dark and light orange arcs show the scaffold N50 and scaffold N90 values. The central light gray spiral shows the cumulative scaffold count with a white line at each order of magnitude. White regions in this area reflect the proportion of Ns in the assembly. The dark vs. light blue area around it shows mean, maximum and minimum GS vs. AT content at 0.1% intervals (Challis et al. 2020)



Contact maps for the primary and alternate genome assembly generated with PretextSnapshot. Contact maps translate proximity of genomic regions in 3-D space to contiguous linear organization. Each cell in the contact map corresponds to sequencing data supporting the linkage (or join) between two of such regions. Scaffolds are separated by black lines and higher density corresponds to higher levels of fragmentation.
Authors
Jonathan Q Richmond, Jimmy A McGuire, Merly Escalona, Mohan P A Marimuthu, Oanh Nguyen, Samuel Sacco, Eric Beraut, Erin Toffelmier, Robert N Fisher, Ian J Wang, H Bradley Shaffer